PCR assembly
Contents
PCR assembly#
Goal: Create the dsDNA that will be the template to create your RNA of interest.
Time: ~ 4 hours
Introduction:#
We need to go from the cheap short single-stranded DNA (ssDNA) oligos we ordered to the dsDNA which will transcribe to create our RNA of interest. To do this we conduct an PCR assembly reaction. In short, our oligos overlap with each other so they can anneal to each other. This creates multiple primed sites for PCR reactions. The complete double-standed DNA (dsDNA) will be completed in the extension steps. Using a construct designed by primerize this normally succeeds in making the dsDNA we want. Check the assembly with a gel confirming the expected length dsDNA is present and checking for side-products. Finally, we need to quantify how much dsDNA we have in order to calculate the amount to use for the transcription step.
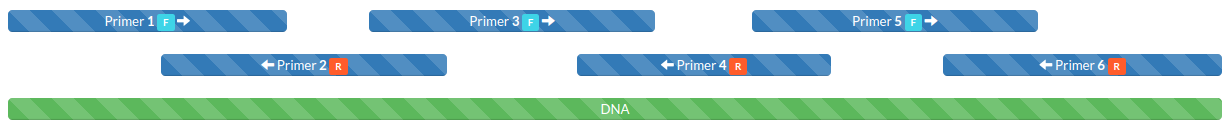
Note
Once you have this complete dsDNA construct, you can amplify it using traditional PCR and the first forward primer and last reverse primer.
Hint
If the assembly fails, first try another polymerase.
Materials:
Oligos from IDT
ddH2O
dNTPs
Polymerase and associated buffer (e.g. Phusion and 5x HF Buffer)
~1.5hr on thermocycler
TBE 1x ~150mL
SYBR Safe
dsDNA ladder
6x Loading dye
Agarose
Gel doc
QIAquick spin columns
Buffer PB*
Buffer PE* or 80% EtOH
Centrifuge
Nanodrop
* recipes available
Method#
1. Primer MasterMix
Check primer concentration (often ~100µM), label primers on cap.
Create a MM with 12.5µM first and last primer (eg 50µL), and 0.125µM others (eg 0.5µL) (eg in 400µL total). You can save MM for future reactions, store at -20°C. [primer MM].
2. Polymerase MasterMix and PCR preparation
Before starting, make sure there is a thermocycler available and reserve with tape.
Each standard reaction is 50µL, recommend 4 standard reactions per assembly. Label PCR tubes for each assembly. Recognize the capacity of your thermocycler, if it is 100µL and you want 4 standard reactions per assembly, each assembly will need to be done in 2 PCR tubes (100µL each, 200µL total).
For each assembly add 16µL of [primer MM] per standard reaction (eg 64µL for 4 standard reactions) directly to the PCR tubes.
Make a Phusion MM for 1.1x the number of reactions you need (number of assemblies x number of standard reactions per assembly). Per reaction you need:
22µL ddH2O
10µL HF-buffer
1µL dNTPs (10mM)
1µL Phusion polymerase add last minute Vortex and spin down.
Add 34µL of Phusion MM per standard reaction to each of your PCR tubes.
Vortex and spin down. Immediately place in thermocycler.
3. PCR cycles
Set the following program on thermocycler (likely saved)
98°C 30sec
35x
[98°C 10sec
64°C 30sec (check annealing temp of oligos, can go a few °C above min Tm)
72°C 30sec] (may need to extend if longer construct, 15-30 per kb)
72°C 10min
12°C inf
4a. Agarose gel
Make agarose gel:
Need 150mL of 1xTBE buffer. Make from 10xTBE and milliQ water.
To 150mL 1xTBE buffer add 10µL SYBR Safe.
Set up a gel box with a ladder and gates. Tape gates down for tighter closure.
Add % mass/volume agarose (eg 4% is 3g, for checking length 4% is ok, if it is quite thick use 3%) in 75mL of the TBE-SYBR Safe.
Microwave sample for around 1.5min, it should look clear and uniform, otherwise microwave 30sec more. Then pour into the gel doc. Use a heat-resistant glove! For low %agarose some may leak under the gates, that is ok.
Let the gel set for ~1hour. Clean glassware you melted gel in after it dries so as not to get agarose in the drain.
Prepare samples:
For PCR-assembly products recommend: 1µL sample, 1µL dye, 4µL ddH2O. In general need dyes are 6x so need to be ⅙ of total.
Choose ladder according to size of product (usually 50bp ladder suffices). If ladder is ready-to-go; do not add ddH2O or dye.
Plan out lanes, note can leave lanes empty. Make sure the ladder(s) reasonable distance to every sample.
Load samples:
Remove gates and wiggle ladder to remove vertically. Fill with remaining TBE-SYBR Safe solutions so the wells are fully flooded.
Pipette 6µL of prepared sample or ladder into wells according to plan.
Attach the wires (match colors!) to the gel box and the power pack. Bubbles should form at electrodes and dyes run down the gel.
Run the gel at 15-20W (eg 150V, 100mA) for ~30min, check progress by using the blue light transilluminator.
Visualize gel:
Once bands are sufficiently discernable, stop the run, take the gel from the buffer and image it on a UV-transilluminator using the UV (SYBR) setting.
Warning
If there are significant side-products you may want to gel-purify the correct sized products, however it may be ok to proceed if you will exclude these products in dPAGE purification of the RNA. If DNA gel reveals multiple bands, excise the band of interest and use the Qiagen QiaQuick Gel Extraction Kit to gel-purify the band before IVT.
4b. E-gel
Prep samples by mixing 15µL E-gel loading buffer with 5µL sample (use 96 well plate)
Mix 5µL E-gel DNA ladder with 15µL loading buffer
Take E-gel cassette out of package and remove comb before placing into E-gel machine
Load water in the outer lanes if there are extra lanes (all lanes must be filled)
Load prepared DNA ladder and samples
Close the lid and run gel on Program 7 for 10 min (for 2% gels)
Warning
If there are significant side-products you may want to gel-purify the correct sized products, however it may be ok to proceed if you will exclude these products in dPAGE purification of the RNA. If DNA gel reveals multiple bands, excise the band of interest and use the Qiagen QiaQuick Gel Extraction Kit to gel-purify the band before IVT.
5. dsDNA column purification
If you have multiple tubes of the same sample you can mix these all together first. If you may have more than 10µg of DNA, split the sample into multiple columns.
Bind DNA to the membrane:
Place QIAquick spin column in 2mL collection tube.
Add 5:1 Buffer PB:PCR sample and vortex. (eg 1000µL PB to 200µL product). (should be yellow).
Add 700µL at a time to the column. Spin at 13,000 for 1min. Discard flow-through. Repeat until all is in the column.
Wash DNA: 4. Add 700µL Buffer PE. Spin at 13,000 for 1min. Discard flow-through. Dap on a chem wipe to remove as much ethanol as possible.
Remove residual ethanol: 5. Spin at 13,000 for 2min. 6. Move column to a well-labeled 1.5mL tube. Dry 15min with an open column.
Elute DNA: 7. Add 30µL ddH2O directly to the membrane. Wait 10min. Spin at 10,000 for 1min. Discard column not flowthrough. 8. Save DNA in -20°C. [dsDNA]
6. dsDNA Quantification
Bring 1µL of sample in PCR tube. If you bring your own pipette to the non-RNAse free nanodrop, wipe it off with RNAse zap before using again.
Wipe down the NanoDrop. Set to dsDNA.
Blank and zero with 1µL ddH2O.
Load 1µL of sample, take measurement.
Check for contaminants. Expect A260/A280~1.8. Do not want a 230 shoulder. Generally, if A260 above 70 needs to dilute.
Write down: A260, A280, and Reported ng/µL.
Calculate concentration: DNA (μM) = [A260 x 50,000] / [660 x length (bp)]
Note
Note, if you have a epPCR primer site at the end of your construct you will want to remove this before transcribing the RNA for cryo-EM, this requires a digest, another column purification, and checking on the gel to see the size has reduced by the expected amount.
7. optional DNA gel purification
If DNA gel reveals multiple bands, you may want to gel purify the DNA of the correct length. Make a 1% agarose gel and load your sample, excise the band of interest and use the Qiagen QiaQuick Gel Extraction Kit to gel-purify the band before IVT.
Image the gel under blue light, mark the band with a sharpie (red).
Weigh an empty 1.5 mL microcentrifuge tube.
Use a clean, sharp razor to excise the DNA fragment. Try not to cut excess gel.
Add the gel slice to the 1.5 mL tube. Reweigh the tube to get the mass of the gel. If the gel slice is >400 mg, use 2 columns.
For >2% agarose gels (we use 4%), add 6 volumes of Buffer QG (100 mg gel ~100 uL).
Incubate the gel slice and Buffer QG at 50C for 10 min. (or until the gel slice has completely dissolved). Vortex the tube every 2-3 min to help dissolve the gel.
Check that the color of the mixture is yellow. If it is orange or violet, add 10 uL 3M sodium acetate, pH 5.0, and mix. (3M sodium acetate from Ann is pH 5.5)
Add 1 gel volume of isopropanol to the sample and mix.
Add the sample in 750 uL volumes to the QIAquick spin column. Spin at 13,000xg for 1 min.
Discard flow-through. Add until all is on column.
If the DNA will be used for IVT, add 500 uL Buffer QG to the column. Spin at 13,000xg for 1 min. Discard flow-through.
Add 750 uL Buffer PE to the column. Spin at 13,000xg for 1 min. Discard flow-through.
Spin at 13,000xg for 1 min to remove residual wash buffer.
Move column to a well-labeled 1.5 mL tube. Dry 15 min with an open column.
Add 30 uL ddH2O to the membrane. Wait 10 min. Spin at 10,000 for 1 min. Discard column not flowthrough.
Save DNA in -20°C [dsDNA]
change edit to copy on gslides!! make 7 own page but link it, and digest, and primer TODO different assembly enzymes